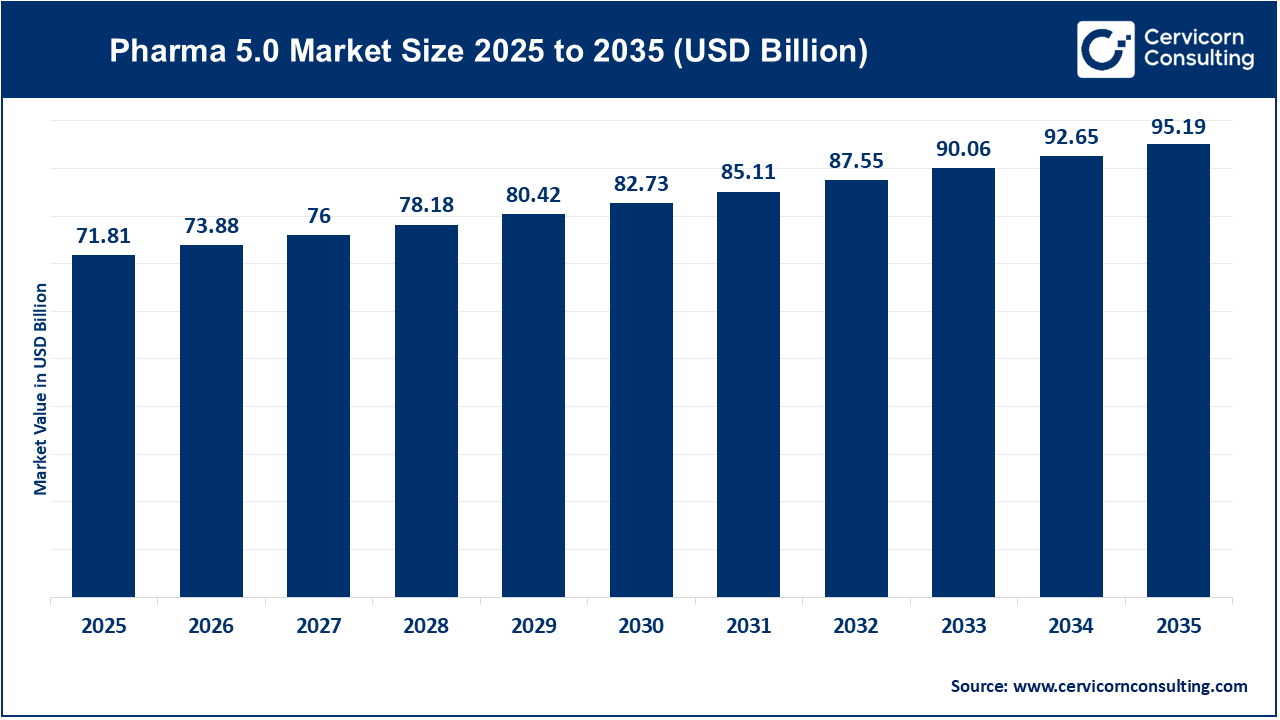
A Comprehensive Guide to FDA Medical Device Regulations
-

- March 19th, 2026
- 1,752 views

FREE SEO Topical Map Generator: Find Your Next Content Ideas
The realm of medical device regulation in the United States is complex, with the fda regulations for medical devices playing a pivotal role in ensuring the safety and efficacy of medical devices. These regulations are designed to protect public health by providing a framework for the evaluation, approval, and monitoring of medical devices throughout their lifecycle. This comprehensive guide delves into the various aspects of FDA Medical Device Regulations, including their history, classification system, approval processes, and post-market surveillance.
History of FDA Medical Device Regulations
The history of FDA Medical Device Regulations dates back to the early 20th century, with significant developments occurring over the decades to address emerging challenges in the medical device industry.
Early Regulations
The regulation of medical devices in the United States began with the Food and Drugs Act of 1906, which aimed to prevent the sale of adulterated or misbranded products. However, this act primarily focused on food and drugs, with limited provisions for medical devices. It wasn't until the Federal Food, Drug, and Cosmetic Act (FDCA) of 1938 that the regulation of medical devices gained more prominence. This act required pre-market approval for new drugs but did not impose similar requirements for medical devices.
Medical Device Amendments of 1976
The Medical Device Amendments of 1976 marked a turning point in the fda regulations for medical devices. These amendments were enacted in response to public health concerns and technological advancements in the medical device industry. The amendments established a regulatory framework for medical devices, introducing a classification system based on the risk associated with each device type. This system categorized devices into three classes: Class I, Class II, and Class III, with Class III devices requiring the most stringent regulatory oversight.
Subsequent Amendments and Reforms
Over the years, several amendments and reforms have been introduced to enhance the fda regulations for medical devices. The Safe Medical Devices Act of 1990 aimed to improve the reporting of adverse events and expand the FDA's authority to fda regulations for medical devices. The Food and Drug Administration Modernization Act of 1997 introduced measures to streamline the regulatory process and facilitate innovation in the medical device industry.
More recently, the 21st Century Cures Act of 2016 introduced provisions to accelerate the development and approval of medical devices while ensuring patient safety. These legislative efforts reflect the evolving nature of FDA Medical Device Regulations and the need to address emerging challenges in the healthcare industry.
Classification of Medical Devices
The classification of medical devices is a fundamental aspect of FDA Medical Device Regulations. The classification system categorizes devices based on their intended use and the risk they pose to patients.
Class I Devices
Class I devices are considered low-risk and are subject to the least regulatory oversight. These devices are typically simple in design and pose minimal risk to patients. Examples of Class I devices include bandages, tongue depressors, and manual surgical instruments. Most Class I devices are exempt from pre-market notification and can be marketed without prior FDA approval. However, manufacturers must adhere to general controls, which include requirements for proper labeling, quality control, and record-keeping.
Class II Devices
Class II devices are considered moderate-risk and require more regulatory oversight than Class I devices. These devices may pose some risk to patients if not used correctly, necessitating additional controls to ensure their safety and effectiveness. Examples of Class II devices include infusion pumps, X-ray machines, and surgical drapes.
To market a Class II device, manufacturers typically need to submit a pre-market notification, also known as a 510(k) submission, to the FDA. The 510(k) submission demonstrates that the device is substantially equivalent to a legally marketed device, known as a predicate device. Substantial equivalence means that the device is as safe and effective as the predicate device and does not raise new questions of safety and effectiveness.
Class III Devices
Class III devices are considered high-risk and require the most stringent regulatory oversight. These devices are often life-sustaining or life-supporting and may pose significant risks to patients. Examples of Class III devices include implantable pacemakers, heart valves, and artificial hearts.
To market a Class III device, manufacturers must obtain pre-market approval (PMA) from the FDA. The PMA process involves a rigorous review of clinical data to ensure the device's safety and effectiveness. Manufacturers must provide evidence from well-controlled clinical trials and submit detailed information about the device's design, manufacturing, and labeling.
The Approval Process for Medical Devices
The approval process for medical devices under FDA Medical Device Regulations varies depending on the device's classification. The primary pathways for approval are the 510(k) process and the pre-market approval (PMA) process.
The 510(k) Process
The 510(k) process is the most common pathway for Class II devices. It allows manufacturers to demonstrate that their device is substantially equivalent to a predicate device. The key steps in the 510(k) process include:
Device Classification: Manufacturers must determine the device's classification and identify a suitable predicate device.
Substantial Equivalence: Manufacturers must demonstrate that their device is as safe and effective as the predicate device. This involves comparing the device's intended use, technological characteristics, and performance testing.
Submission: Manufacturers must submit a 510(k) application to the FDA, including detailed information about the device, its intended use, and supporting data.
FDA Review: The FDA reviews the 510(k) submission to determine if the device is substantially equivalent to the predicate device. The review process typically takes 90 days, although the timeline may vary based on the complexity of the device and the quality of the submission.
Clearance: If the FDA determines that the device is substantially equivalent, it issues a 510(k) clearance, allowing the manufacturer to market the device.
The Pre-Market Approval (PMA) Process
The PMA process is required for Class III devices and involves a more rigorous evaluation of the device's safety and effectiveness. The key steps in the PMA process include:
Clinical Trials: Manufacturers must conduct well-controlled clinical trials to gather evidence of the device's safety and effectiveness. The trials must comply with FDA regulations and may require an investigational device exemption (IDE) to conduct.
PMA Submission: Manufacturers must submit a PMA application to the FDA, including detailed information about the device, clinical trial data, manufacturing processes, and labeling.
FDA Review: The FDA conducts a thorough review of the PMA submission, assessing the device's safety, effectiveness, and quality control measures. The review process typically takes 180 days, although it may take longer for complex devices or applications with incomplete data.
Advisory Committee Review: In some cases, the FDA may seek input from an advisory committee composed of experts in the relevant field. The committee provides recommendations on the device's safety and effectiveness.
Approval: If the FDA determines that the device is safe and effective, it grants pre-market approval, allowing the manufacturer to market the device. The approval may include conditions or post-market requirements to ensure ongoing safety and effectiveness.
Special Pathways and Expedited Programs
In addition to the standard approval processes, the FDA offers special pathways and expedited programs to facilitate the development and approval of certain medical devices.
De Novo Classification
The De Novo classification process provides a pathway for novel devices that do not have a suitable predicate device. This process is intended for devices that are considered low to moderate risk but do not fit into the existing classification framework. The De Novo process involves a risk-based evaluation to determine the appropriate classification and regulatory controls for the device.
Humanitarian Device Exemption (HDE)
The Humanitarian Device Exemption (HDE) program is designed for devices intended to treat or diagnose rare diseases or conditions affecting fewer than 8,000 individuals in the United States per year. The HDE process allows manufacturers to obtain marketing approval without demonstrating effectiveness, provided the device does not pose an unreasonable risk to patients. However, manufacturers must still demonstrate the device's safety and provide evidence of probable benefit.
Breakthrough Devices Program
The Breakthrough Devices Program is designed to expedite the development and approval of devices that address unmet medical needs or offer significant advantages over existing treatments. The program provides manufacturers with priority review, interactive communication with the FDA, and access to senior agency personnel. Devices that qualify for the Breakthrough Devices Program receive accelerated review and approval, allowing them to reach the market more quickly.
Post-Market Surveillance and Compliance
Once a medical device is approved and marketed, it is subject to ongoing monitoring and compliance requirements under FDA Medical Device Regulations.
Post-Market Surveillance
Post-market surveillance is a critical component of the FDA's regulatory framework, ensuring the continued safety and effectiveness of medical devices. Manufacturers are required to report adverse events, conduct post-market studies, and implement corrective actions as needed.
Medical Device Reporting (MDR): Manufacturers, importers, and user facilities must report adverse events and device malfunctions to the FDA. The MDR system helps identify potential safety issues and trends that may require further investigation.
Post-Approval Studies: For certain high-risk devices, the FDA may require post-approval studies to gather additional data on the device's safety and effectiveness in real-world settings. These studies help ensure that the device performs as expected and that any long-term risks are identified and addressed.
Recalls and Corrective Actions: If a device is found to pose a risk to patients, the FDA may initiate a recall or require the manufacturer to implement corrective actions. Recalls can range from minor labeling changes to the removal of the device from the market.
Compliance and Enforcement
The FDA has the authority to enforce compliance with FDA Medical Device Regulations and take action against manufacturers that violate regulatory requirements.
Inspections: The FDA conducts routine inspections of manufacturing facilities to ensure compliance with quality system regulations. Inspections may be conducted periodically or in response to specific concerns.
Warning Letters: If the FDA identifies violations during an inspection or review, it may issue a warning letter to the manufacturer. The letter outlines the violations and requests corrective actions to address the issues.
Civil and Criminal Penalties: In cases of significant violations, the FDA may pursue civil or criminal penalties against manufacturers. Penalties can include fines, injunctions, or criminal charges, depending on the severity of the violation.
Global Harmonization and Future Trends
As the medical device industry continues to evolve, the FDA is working to harmonize its regulations with international standards and address emerging trends in healthcare.
International Harmonization
The FDA is actively involved in global harmonization efforts to align its regulations with international standards. The International Medical Device Regulators Forum (IMDRF) and the Global Harmonization Task Force (GHTF) are two key initiatives aimed at promoting consistent regulatory practices worldwide. By aligning with international standards, the FDA seeks to facilitate global market access for medical devices and reduce regulatory burdens for manufacturers.
Emerging Trends
Several emerging trends are shaping the future of FDA Medical Device Regulations, including advancements in digital health, personalized medicine, and cybersecurity.
Digital Health: The rise of digital health technologies, such as mobile medical apps and wearable devices, presents new challenges and opportunities for regulation. The FDA is developing guidelines to address the unique aspects of digital health products, including software validation, data security, and interoperability.
Personalized Medicine: Personalized medicine involves tailoring medical treatments to individual patients based on their genetic, environmental, and lifestyle factors. The FDA is exploring regulatory approaches to support the development of personalized medical devices and ensure their safety and effectiveness.
Cybersecurity: As medical devices become more connected and integrated with healthcare networks, cybersecurity is a growing concern. The FDA is working to establish cybersecurity guidelines and best practices to protect patient data and ensure the integrity of medical devices.
Conclusion
Navigating the landscape of fda regulations for medical devices requires a deep understanding of the regulatory framework, classification system, approval processes, and post-market requirements. By adhering to these regulations, manufacturers can ensure the safety and effectiveness of their devices while fostering innovation and addressing emerging healthcare challenges. As the industry continues to evolve, the fda regulations for medical devices remains committed to adapting its regulations to keep pace with technological advancements and global harmonization efforts, ultimately protecting public health and improving patient outcomes.